With the increasing availability of modulator therapies for people with cystic fibrosis, end-stage infection with Pseudomonas aeruginosa is becoming less common. But are we going to see a rise in the prevalence of the early cystic fibrosis lung pathogen Haemophilus influenzae?
The problem: cystic fibrosis
The life expectancy of people with cystic fibrosis (CF) has made huge advancements over the last few decades. What was once a life expectancy of 4-5 years is now 49 years in the United Kingdom, and is expected to keep increasing. As the most common recessive genetic disorder worldwide, CF currently affects over 160,000 individuals and is largely attributable to defects in one gene, the cystic fibrosis transmembrane conductance regulator (CFTR). This codes for a chloride ion channel protein, responsible for regulating water homeostasis across the epithelium of multiple organs.
Although a multi-organ disease, CF primarily manifests within the lungs, as the disrupted water regulation results in the accumulation of viscous mucus on the surface of epithelial cells in the airways, creating an excellent breeding ground for bacterial infection. Cilia lining the surface of epithelial cells are unable to beat away bacteria entrapped within the mucus due to its high viscosity, which leads to chronic bacterial infection, a perpetuating inflammatory cycle, and severe respiratory complications.
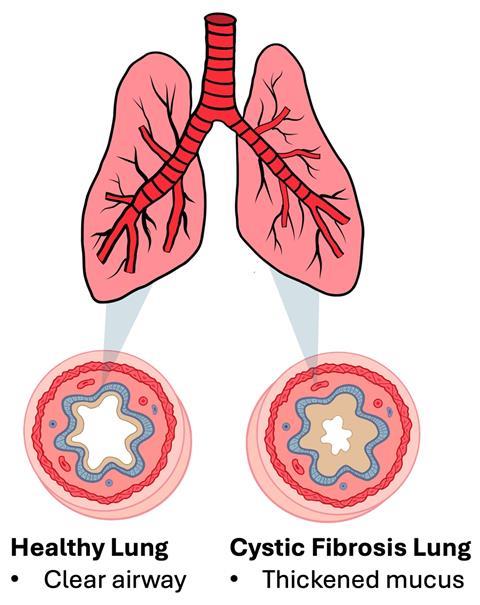
Although attributable to one gene, over ~2000 mutations have been identified in CFTR, with ~700 of these resulting in cystic fibrosis. As such, there is currently no cure for CF, but the prognosis is continually improving due to a multi-disciplinary approach to care, treatment advances, newborn screening, and the development of proteins to help correct the function of the CFTR, known as modulator therapies.
“Classic” course of lung infection
Respiratory failure caused by chronic infections remains one of the main causes of mortality, with several pathogens known to colonise the lungs of people with CF. The classic course of infection commonly begins with Haemophilus influenzae and Staphylococcus aureus in infancy, followed by Pseudomonas aeruginosa in teenage years. Prevalence of both H. influenzae and S. aureus drops off in later years as P. aeruginosa dominates.
As the dominant end-stage pathogen, there is a plethora of research on P. aeruginosa lung infection, ranging from investigating virulence factors, modelling infection dynamics, and the development of novel treatments. Yet, the knowledge of H. influenzae in a CF lung environment is relatively understudied, with the majority of work undertaken in standard laboratory media, which is likely due to how challenging H. influenzae can be to culture in more minimal media compared to other CF pathogens. However, this means the current knowledge of H. influenzae in CF lung infection is largely attributable to experiments performed in media that are not representative of a CF lung environment.
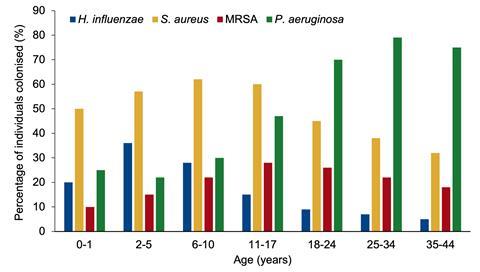
You may be thinking, why would we study an early pathogen that does not cause major issues for people with CF? If we investigate the pathogen prevalence graph using data from the CF Foundation Patient Registry Report, the ages at which H. influenzae prevalence decreases are where P. aeruginosa infection increases. So, is H. influenzae influencing the colonisation of the more severe P. aeruginosa in some way? Further, how might CFTR modulator therapy change this “classic” infection progression?
The unknown interactions between H. influenzae and P. aeruginosa
Very little is known about the interactions between H. influenzae and P. aeruginosa, especially in a CF-mimicking environment. It has been hypothesized that damage to the mucosal epithelium caused by initial H. influenzae infection facilitates the colonisation of P. aeruginosa. As such, people with CF often undergo extensive antibiotic treatment to clear H. influenzae infection. However, some recent work by Lindgren et al. has shown that prior H. influenzae lung colonisation actually reduces the inflammatory response and lung damage caused by P. aeruginosa in a mouse model, compared with infection by P. aeruginosa alone. This suggests the aggressive antibiotic treatment of H. influenzae may allow P. aeruginosa to colonise more easily due to reduced competition, and as a result, induce greater inflammation and lung damage.
So, should we allow H. influenzae, a less severe pathogen, to colonise the lungs of people with CF, to prevent or reduce the severity of an often worse, P. aeruginosa infection? As little is known, a greater understanding of the interactions between early CF pathogens and P. aeruginosa is needed to shape treatment guidelines and improve the quality of life of people with CF.
Modulator therapies: changing the landscape of CF lung infection
In 2012, we saw the approval of the first modulator therapy for CF, a revolutionary treatment advancement effective in ~90% of CF patients. Modulator therapies are small molecules that restore, or partially restore, the function of the CFTR protein. Currently licensed modulators are ivacaftor, lumacaftor, tezacaftor, and elexacaftor, and the cutting-edge combinations of ivacaftor, tezacaftor, and elexacaftor (TRIKAFTA) as well as vanzacaftor, tezacaftor, and deutivacaftor (ALYFTREK), which was approved in children 6 years and older as recently as December 2024. Modulator therapies have been demonstrated to positively influence lung function and reduce pulmonary exacerbations, and their introduction is thought to increase life expectancy above 50 years. However, the long-term effects of modulator therapies remain unknown, and the effect of modulator therapies on bacterial infection remains even more elusive.
Ivacaftor was the first modulator therapy, developed for people with a G551D mutation in the CFTR (about 4% of people with CF). It has been shown to reduce the prevalence of P. aeruginosa over the course of both 5 and 6 years in two different studies, with smaller reductions seen in S. aureus. The effect of ivacaftor on H. influenzae is less studied, but the odds of culture positivity have been observed to increase after 1 year in a study with 151 individuals taking ivacaftor, compared to P. aeruginosa, which decreased in respiratory cultures. A comparison of data from CF registries in the United Kingdom and United States showed no change in H. influenzae prevalence after 2 years of ivacaftor, compared to the decrease seen with P. aeruginosa and S. aureus, and a similar study highlighted that whilst a 30% decrease in P. aeruginosa was seen with ivacaftor, the relative proportion of H. influenzae increased up to 80%.
Even with the TRIKAFTA combination therapy, similar results have been seen. The prevalence of P. aeruginosa and S. aureus decreased one year after TRIKAFTA treatment, but the density of H. influenzae increased. Another study highlighted that 14 and 50 weeks after starting TRIKAFTA, individuals who were H. influenzae negative before treatment now had increased H. influenzae DNA levels in deep cough swabs.
It has been hypothesised that the reduction of P. aeruginosa in the presence of modulator therapies is what allows H. influenzae to thrive, due to the lack of P. aeruginosa pyocins inhibiting H. influenzae growth. As such, this poses the question: will H. influenzae emerge as a new pathogen in people with CF, with the increasing availability of modulator therapies, and people with CF less likely to reach “classic” end-stage infection with P. aeruginosa? If this is the case, we need to be ready to tackle an increasing number of CF patients with H. influenzae infection by gaining a greater understanding of its growth profiles, antimicrobial susceptibility, and virulence in the CF lung.
H. influenzae grows poorly in published formulations of synthetic CF sputum media
A great way to study CF pathogens is through the use of artificial sputum media, which have been developed to mimic the in vivo nutritional composition of CF sputum. These media are designed to cue similar bacterial growth, pathogenicity, and gene expression patterns as are seen in vivo, increasing the clinical relevance of laboratory research and aiding the discovery of effective antibacterial therapies. Several lineages of artificial sputum exist, containing mainly mucin, DNA, amino acids, and a source of lipid. However, each synthetic CF sputum recipe was developed and optimised to support P. aeruginosa growth, and when I started my PhD in 2021, there was no published literature for H. influenzae growth in artificial sputum media.
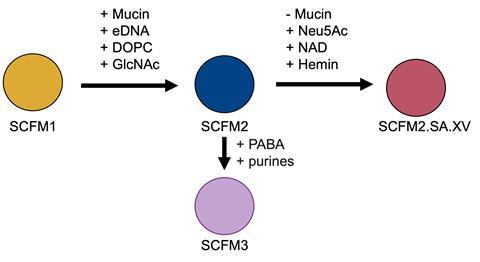
Through my PhD, I investigated the growth of laboratory H. influenzae strains in several formulations of SCFM, including SCFM1, SCFM2, and SCFM3. SCFM1, which is widely used to study P. aeruginosa infection in vitro, struggled to support the growth of H. influenzae. I found that replacing the whole mucin in SCFM2 with just the sialic acid moiety of mucin improved H. influenzae growth, and supplementation with NAD and hemin was required for the growth of H. influenzae. This suggests a source of these two molecules is present in the lungs of people with CF, which may arise from the presence of blood in sputum, which is common in ~60% of people with CF due to airway inflammation. In SCFM3, NAD is present, and hemin has been determined as ‘potentially’ available, which also suggests their likely presence in the CF lung. Finally, I found that the presence of extracellular DNA in my modified SCFM2 was especially important in significantly enhancing the growth and biofilm formation of H. influenzae.
With several published variants of SCFM struggling to support the growth H. influenzae, a common CF pathogen, this poses the question of really, how representative SCFM is of the in vivo lung environment? My research has already suggested that the presence of NAD, hemin, and extracellular DNA are important components to promote H. influenzae growth when utilising SCFM. But further, that lung homogenate (utilising bead-beaten bronchiole tissue from pig lungs) combined with modified SCFM promoted the growth of H. influenzae further. This suggests there are components in the lung tissue that promote bacterial growth, and that SCFM can be optimised further for usage with H. influenzae following potential proteomic analysis of the components within lung homogenate.
Increasing antimicrobial resistance
Several studies have investigated the antibiotic susceptibility profiles of common pathogens in SCFM and in laboratory media, finding staggering differences in antibiotic susceptibility profiles. In the case of P. aeruginosa, colistin tolerance was increased when grown in SCFM over standard laboratory media. By performing research in host-mimicking conditions, we can gain a more realistic understanding of the infection dynamics and resistance profiles of important pathogens. Although H. influenzae remains susceptible to many antibiotics, antimicrobial resistance of H. influenzae is on the rise, with resistance to ampicillin common due to the production of a beta-lactamase. The global prevalence of beta-lactamase H. influenzae and multidrug-resistant H. influenzae has been determined as 24.9% and 23.1%, respectively.
Almost all antimicrobial susceptibility assays with H. influenzae are performed on chocolate agar or supplemented brain heart infusion broth. However, the conditions of the CF lung are known to promote antibiotic tolerance, not only due to the increased biofilm formation – which can increase antibiotic tolerance as much as 1000-fold relative to planktonic cultures – but also because the hypoxic conditions of the viscous CF lung mucus can alter bacterial metabolism and render antibiotics targeting these mechanisms of action ineffective. With the potential of H. influenzae to become an emerging pathogen, it is crucial there is more research on H. influenzae in host-mimicking conditions to inform antibiotic susceptibility and improve patient outcomes.
Conclusion
Although the increasing availability of modulator therapies is improving CF lung function, there is increasing evidence that P. aeruginosa and S. aureus prevalence is decreasing, whilst H. influenzae prevalence is increasing. It is likely that people with CF will see H. influenzae as an emerging pathogen, and with currently very little research into H. influenzae in a CF lung environment, more research needs to be done to understand its antibiotic susceptibility profiles, biofilm formation, interactions with other CF pathogens, and ultimately, what role H. influenzae has in cystic fibrosis lung infection. To research this, further optimisation of SCFM is required to support H. influenzae growth, and potentially, through analysis of what lung tissue contributes to the nutrient pool utilised by H. influenzae.





No comments yet