Legionnaires’ disease (LD), a rare and severe type of pneumonia, is a respiratory infection caused by species of Legionella bacteria.
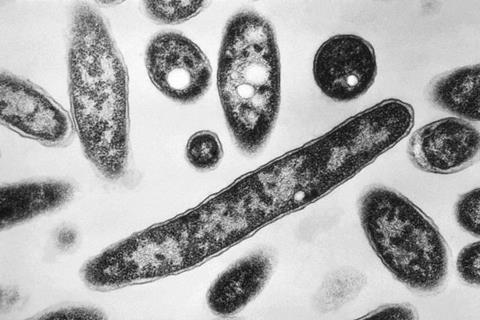
One of the most accurate ways to diagnose LD is to perform culture on samples from a patient’s lower respiratory tract, but those samples are difficult to obtain. In addition, growing cultures requires specialized culture media and incubation times and conditions that many laboratories lack; as a result, LD is likely underdiagnosed.
This week in Journal of Clinical Microbiology, a journal of the American Society for Microbiology, researchers from the New York State Department of Health in Albany describe a cost-effective approach for using whole genome sequencing to identify L. pneumophila that doesn’t require culturing. The researchers say the method could be used to better analyze available specimens and enable closer surveillance of LD outbreaks, especially in places or cases where culturing is difficult.
Cultured isolate
“It’s challenging when there’s an outbreak and we have some samples, but we can’t get a cultured isolate out,” said Kimberlee Musser, Ph.D., Chief of Bacterial Disease at the New York State Department of Health. The new method doesn’t require growing that isolate, she said. “We only rely on the organism that’s in the specimen.”
The approach relies on a technique called RNA baiting. It begins with known organisms for which researchers already have cultures. “We chop them up into small pieces,” Musser said, and then generate RNA associated with those species. They use those RNA strands—the baits—to extract DNA from the unknown specimen, which can then be sequenced and confirmed.
Their approach, called hybridization capture, has already proved useful. One of the deadliest LD outbreaks in U.S. history occurred in 2015, in a low-income community in the South Bronx in New York City. The outbreak killed 16 people and hospitalized more than 100 others. The investigation led researchers, including Musser, to aerosols from a particular cooling tower in the area as the source.
Hybridization capture
However, not all the cases were clearly linked to the culpable strain using whole genome sequencing. In the new paper, Musser and her colleagues used hybridization capture on autopsy samples (that could not be cultured) from victims of the 2015 outbreak and were able to identify the outbreak source in some cases. “[Autopsy] samples are often some of the hardest to culture, so having a method that we can use to sequence these samples is very useful,” said Phil Weeber, a research scientist at the New York Department of Health and one of three first authors on the paper.
“We wanted to be able to utilize the new method without having to have the cultured isolate,” Musser said. “We’re pulling out just the information we want. It’s really giving us pieces of the puzzle that we didn’t have previously.”
In previous work, Musser’s team used hybridization capture to identify Escherichia coli that produce Shiga toxin, using stool samples and without requiring cultures. Hybridization capture, she said, offers a way to improve infectious disease surveillance.
“We’re in a very exciting time with these new next-generation sequencing methods, and I think the more work we can do to better understand and utilize lots of methods, the closer we get to being able to answer many questions during public health investigations,” Musser said.




No comments yet