Viral infectious diseases pose significant challenges due to the rapid evolution of viruses through mutations. This was particularly evident during the COVID-19 pandemic, when emerging variants of SARS-CoV-2 sparked new waves of infection. These variants often carry mutations that made them more transmissible, allowing them to spread rapidly across populations.
Understanding a virus’s “fitness”—its ability to spread in a host population—has become essential for managing and anticipating viral threats. Though there are methods to assess the fitness of variants based on mutation patterns, statistical models that consider interactions between mutations are lacking.
READ MORE: Seemingly ‘broken’ genes in coronaviruses may be essential for viral survival
READ MORE: First human respiratory organoid culture system reveals secret of Omicron’s transmissibility
To address this challenge, a team of researchers led by Associate Professor Jumpei Ito, including Dr. Adam Strange and Professor Kei Sato, from The Institute of Medical Science at The University of Tokyo, Japan, introduced CoVFit, a novel framework designed to predict the fitness of SARS-CoV-2 variants. Their findings were published online in Nature Communications on May 13, 2025.
CoVFit integrates molecular data with large-scale epidemiological data to provide a predictive model that helps us understand why some variants succeed while others do not. This framework offers more than just tracking the spread of the virus; it reveals the underlying reasons for its success, making it a powerful tool for real-time surveillance and response in the face of ongoing and future viral outbreaks.
Spike protein
The CoVFit model was developed through an innovative approach that combined molecular and epidemiological data. The team focused on mutations in the spike (S) protein, which affect the virus’s ability to escape immune protection from past infections or vaccinations, alongside population-level trends like variant prevalence over time and in different regions. By combining this information, CoVFit was trained and tested to predict a variant’s fitness score.
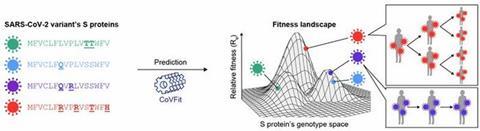
Dr. Ito explains: “We developed an artificial intelligence (AI) model, CoVFit, that predicts the fitness of SARS-CoV-2 variants based on the S protein sequence. Using CoVFit, we elucidated which mutations SARS-CoV-2 has acquired to enhance its fitness and repeatedly expand its spread.”
The model demonstrated an impressive ability to predict the evolutionary impact of single amino acid substitutions in the virus with high accuracy, offering insights into how the virus evolves and spreads. Dr. Ito also notes: “CoVFit is expected to enable the early detection of high-risk variants with a high potential for widespread transmission, ideally at the point when just a single sequence of the variant is registered in a database.”
Forecasting viral evolution
The team further developed a prospective approach to forecast viral evolution using CoVFit. They systematically generated in silico mutant variants by introducing all possible single amino acid substitutions into a reference strain and predicted the fitness of each. This enabled the identification of mutations with a high likelihood of emerging in future variants.
When applied to the Omicron BA.2.86 lineage, CoVFit predicted that substitutions at S protein positions 346, 455, and 456 would enhance viral fitness. Remarkably, these exact mutations were later observed in BA.2.86 descendant lineages—JN.1, KP.2, and KP.3—that subsequently spread globally. Dr. Ito concludes: “These findings underscore CoVFit’s ability to anticipate evolutionary changes driven by single amino acid substitutions.”
In conclusion, CoVFit represents a major breakthrough in our ability to predict, interpret, and respond to viral evolution. By integrating molecular biology with population-level data through AI, it provides a flexible, transparent, and timely approach to pandemic preparedness. As viruses continue to evolve, tools like CoVFit will play a critical role in guiding proactive and informed public health responses worldwide.
Topics
- Adam Strange
- AI & in silico microbiology
- AI & in silico microbiology
- Artificial Intelligence & Machine Learning
- Asia & Oceania
- CoVFit
- COVID-19
- Emerging Threats & Epidemiology
- Epidemiology
- fitness
- Innovation News
- Jumpei Ito
- Kei Sato
- Omicron BA.2.86
- One Health
- SARS-CoV-2
- The University of Tokyo
- variants
- viral evolution




.jpg){kind=link}
No comments yet